| .. | ||
| assets | ||
| setup | ||
| README.md | ||
| scRNA_analysis_preprocessing.ipynb | ||
START HERE with the Single-Cell Analytics Playbook
Table of Contents
Get Started Now!
The scRNA Analysis Preprocessing Notebook is an end to end GPU-accelerated single-cell analysis workflow using RAPIDS-singlecell, a GPU accelerated library developed by scverse®. In this notebook, we understand the cells, run ETL on the data set then visiualize and explore the results. It should take less than 3 minutes to complete the workflow.
CLICK HERE TO BEGIN
Using the DGX Spark can help you easily GPU Accelerate your Data Science and Machine Learning based workflows using the RAPIDS Open Source ecosystem so that you can go from data to information to insights faster than ever before!


Deep Dive
This Notebook is for those who are new to doing basic analysis for single cell data, as the end to end analysis of is the best place to start, where you are walked through the steps of data preprocessing, quality control (QC) and cleanup, visualization, and investigation. Let's deep dive the process!
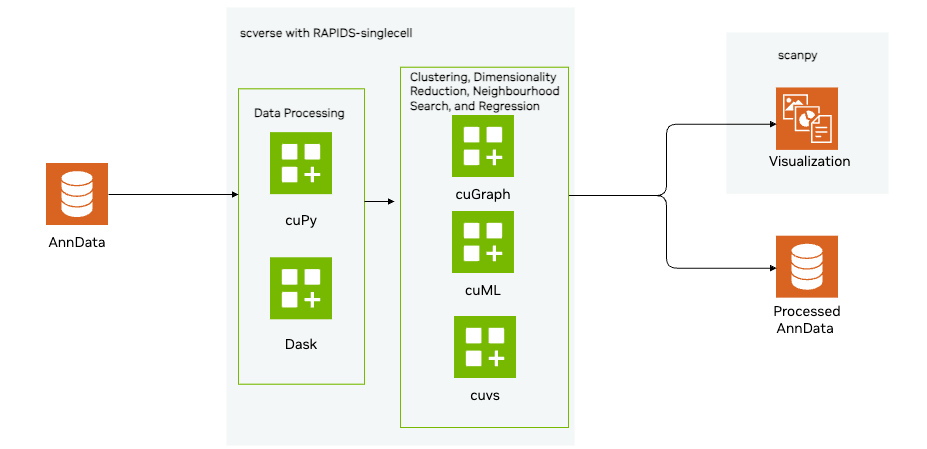
-
Load and Preprocess the data
- Load a sparse matrix in h5ad format using Scanpy
- Preprocess the data, implementing standard QC metrics to assess cell and gene quality per cell, as well as per gen
-
QC cells visually to understand the data
- Users will learn how to visually inspect 5 different plots that help reflect quality control metrics for single cell data to:
- Identify stressed or dying cells undergoing apoptosis
- Empty droplets or dead cells
- Cells with abnormal gene counts
- Low quality or overly dominant cells
- Users will learn how to visually inspect 5 different plots that help reflect quality control metrics for single cell data to:
-
Filter unusual cells
- Users will learn how to remove cells with an extreme number of genes expressed
- Users will filter out cells with an unusual amount of mitochondrial content
-
Remove unwanted sources of variation
- Select most variable genes to better inform analysis and increase computational efficiency
- Regress out additional technical variation that we observed in the visual plots (Note, this can actually remove biologically relevant information, and would need to be carefully considered with a more complex data set)
- Standardize by using a z-score transformation
-
Cluster and visualize data
- Implement PCA to reduce computational complexity. We use the GPU-accelerated PCA implementation from cuML, which significantly speeds up computation compared to CPU-based methods.
- Identify batch effects visually by generating a UMAP plot with graph-based clustering
-
Batch Correction and analysis
- Remove assay-specific batch effects using Harmony
- Re-compute the k-nearest neighbors graph and visualize using the UMAP.
- Perform graph-based clustering
- Visualize using other methods (tSNE)
-
Explore the biological information from the data
- Differential expression analysis: Identifying marker genes for cell types
- Implement logistic regression
- Rank genes that distinguish cell types
- Trajectory analysis
- Implement a diffusion map to understand the progress of cell types
- Differential expression analysis: Identifying marker genes for cell types
These notebooks will be valuable for single-cell scientists who want to quickly evaluate ease of use as well as explore the biological interpretability of RAPIDS-singlecell results. Secondarily, scientists will find value in learning to apply these methods to very large data sets. This repository is also broadly useful for any data scientist or developer who wants to run and evaluate single cell methods leveraging RAPIDS-singlecell. Data sets used for this tutorial were made publicly available by 10X as well as CZ cellxgene.
If you like this notebook and the GPU accelerated capability, please do these two things:
- Explore the rest of the single cell notebooks, through the Single Cell Analysis AI Blueprint
- Support scverse's efforts by please learn more about them here as well as consider joining their community.
Directory Structure
- scRNA_analysis_preprocessing.ipynb - Main playbook notebook
START_HERE.md- Quick Start Guide to the Playbook Environment. It will also be found in the main directory of the Jupyter Lab. Please start there!cuDF, cuML, and cuGraph folders- more example notebooks to continue your GPU Accelerated Data Science Journey.
DIY Installation
If you like what you see and want to run more GPU accelerated for Genomics, BioInfomatics, or Single Cell research, please do the following
pip install -r ./setup/requirements.txt
Inside this requirements file, are some pinned versions of all the libraries needed. When pinned, it will only download that specific version, which will ensure that you have stablity as a product. When unpinned, pip or uv will download the latest versions where everything should work. If you're planning to upgrade to the lastest technological stack, you should unpin the libaries, however your mileage may vary.
Support
If you have any questions about these notebooks or need support, please open an Issue on the Single Cell Analysis AI Blueprint repository and we will respond there.